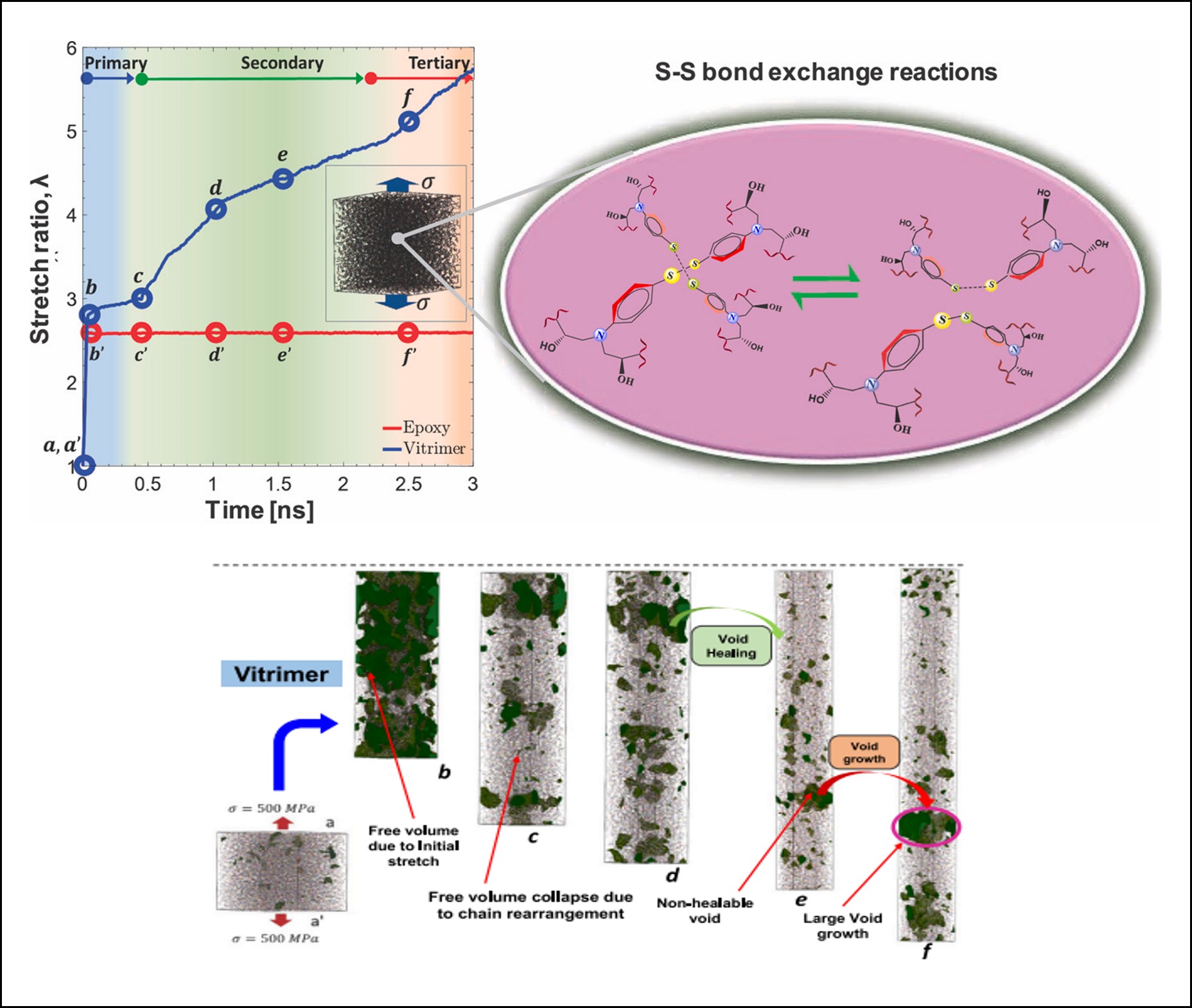
Development of MD Framework for Dynamic Polymer Networks: Created a topology-based reaction scheme to model dynamic disulfide bond exchange in vitrimers under mechanical load, enabling atomic-scale analysis of creep.
Unlike ReaxFF methods, which simulate full reaction pathways at quantum-mechanical resolution (incurring high computational costs), this scheme avoids explicit energy barrier calculations by focusing on topological eligibility rules, making it 10–100x faster for creep simulations under mechanical load. We have identified three-stage creep dynamics (primary/secondary/tertiary) in disulfide vitrimers driven by bond reorientation. In the primary creep regime, elastic/viscoelastic strain dominated by chain alignment is seen. During secondary creep, steady-state strain rate from bond exchange orthogonal to loading axis is seen, reducing stiffness compared to traditional non-recyclable epoxies. We also observed a unique bond alignment mechanism during secondary creep.
This research directly advances IRG 2’s mission to design covalent adaptable networks (CANs) with tailored responsiveness. The simulations reveal how bond reorientation governs vitrimer creep—a critical challenge for recyclable plastics and self-healing composites targeted by IRG 2. We propose that chemistry changes or additives that can prevent the realignment of dynamic bonds can be an effective strategy to mitigate creep in vitrimers.
Published: Singh G, Varshney V, Sundararaghavan V. Understanding creep in vitrimers: Insights from molecular dynamics simulations. Polymer. 2024 Nov 15;313:127667. https://doi.org/10.1016/j.polymer.2024.127667